

Reproduction Biology
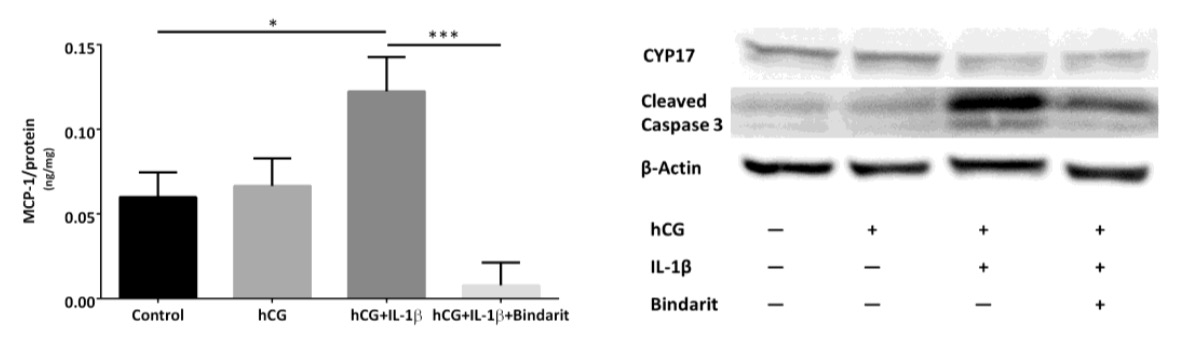
MCP-1 inhibition of mouse Leydig cells: MLTC-1 cells were treated with or without IL-1ß and/or Bindarit for 24h. Secreted MCP-1 was measured by ELISA (left figure). The cell extracts were separated by SDS-PAGE and transferred on a membrane by Western Blotting. Subsequently changes of CYP17 and cleaved caspase 3 were detected (right figure).
The Metabolic Syndrome (MetS) is diagnosed as an accumulation of hypertension, increase of waist circumference, dyslipidemia, and increased blood glucose levels and leads to hypogonadism and male subfertility, reportedly. However, inherent mechanisms are under discussion. Inflammation through infections contributes to nearly 15% of total cases of human infertility. As emerging evidence shows MetS leads to reduced male fertility, the role of inflammation in the MetS-related male subfertility needs to be investigated, and dysregulation of certain pro-inflammatory factor(s) could be held responsible for the failed reproductivity of MetS-stressed subjects. In this project, leptin-resistant mice were used as a model to detect signs of testicular inflammation and impaired Leydig cell function. Mouse and cell models were adopted to study Leydig cells and epididymal sperm in the context of full MetS syndrome. Male BKS(D)-Leprdb+/+/JOrlRj (db/db) mice exhibited MetS and testicular dysfunction after the age of 6 weeks. They were characterized by small testis, low testosterone level, and impaired Leydig cell function. Inflammation was present in the testis of db/db mice, as indicated by upregulated IL-1β and MCP-1 gene expression. Interestingly, the ratio of M1 to M2 macrophages was reduced while testicular corticosterone concentrations were enhanced in db/db mice as opposed to wildtype controls. In vitro treatment of mouse Leydig cells with IL-1β enhanced MCP-1 secretion along with increased caspase 3 expression that is activated in the cell as an indicator for apoptosis. Leydig cell function was rescued by chemical inhibition of MCP-1. Meanwhile, reduced expression of ATF6 indicates involvement of ER stress in the progression of subfertility in db/db mice. Collectively, these findings suggest that the inflammatory environment and ER stress in testis are partially responsible for the damaged fertility in mice with MetS. Considering the critical role of MCP-1, inhibition of MCP-1 may be a potential target for therapeutic intervention in treatment of MetS associated subfertility.
Hyperglycemia and male infertility (Maresch C et al., Human Reproduction Update, 2018). To date several reviews have addressed the issue of diabetes-related male infertility but most have focused on how metabolic syndrome causes the decline in male fertility. However, a comprehensive overview as to how diabetes-induced hyperglycemia impairs male fertility is missing. Impaired regulation of glucose and the resultant hyperglycemia are major threats to the health of individuals in modern societies especially given the rapidly rising prevalence affecting an increasing number of men in their reproductive years. Consequently, diabetes-induced hyperglycemia is likely to contribute to a decline in global birth rates especially in those societies with a high diabetic prevalence.
We performed a systematic review on the literature reporting on the impact of hyperglycemia on male reproductive health with a particular emphasis on the molecular mechanisms that influence the testis and other parts of the male reproductive tract.
A systematic search of the literature published in the MEDLINE-Pubmed database (http://www.ncbi.nlm.nih.gov/pubmed) and Cochrane Library (http://www.cochranelibrary.com) was performed, as well as hand searching reference lists, from the earliest available online indexing year until May 2017, using diabetes- and male fertility-related keywords in combination with other search phrases relevant to the topic of hyperglycemia. Inclusion criteria were: clinical studies on type 1 diabetic (T1D) men and studies on T1D animal models with a focus on reproductive parameters. Case reports/series, observational studies and clinical trials were included. Studies on patients with type 2 diabetes (T2D) or animal models of T2D were excluded to distinguish hyperglycemia from other metabolic effects.
A total of 890 articles were identified of which 197 (32 clinical, 165 animal studies) were selected for qualitative analysis. While the clinical data from men with hyperglycemia-induced reproductive dysfunction were reported in most studies on T1D, the study designs were variable and lacked complete information on patients. Moreover, only a few studies (and mostly animal studies) addressed the underlying mechanisms of how hyperglycemia induces infertility. Potential causes included impaired function of the hypothalamic-pituitary-gonadal axis, increased DNA damage, perturbations in the system of advanced glycation endproducts and their receptor, oxidative stress, increased endoplasmatic reticulum stress, modulation of cellular pathways, impaired mitochondrial function and disrupted sympathetic innervation. However, intervention studies to identify and confirm the pathological mechanisms were missing: data that are essential in understanding these interactions. While the effects of regulating the hyperglycemia by the use of insulin and other modulators of glucose metabolism have been reported, more clinical trials providing high quality evidence and specifically addressing the beneficial effects on male reproduction are required. We conclude that interventions using insulin to restore normoglycemia should be a feasible approach to assess the proposed underlying mechanisms of infertility.